Genome Alignment Service¶
Overview¶
The PATRIC Genome Alignment Service uses progressiveMauve to produce a whole genome alignment of two or more genomes. The resulting alignment can be visualized within the PATRIC website, providing insight into homologous regions and changes due to DNA recombination. It should be noted that this service is currently released as beta. As always, we appreciate your feedback.
See also¶
Using the Genome Alignment Service¶
The Genome Alignment submenu option under the Services main menu (Genomics category) opens the Genome Alignment Service input form (shown below).
Options¶
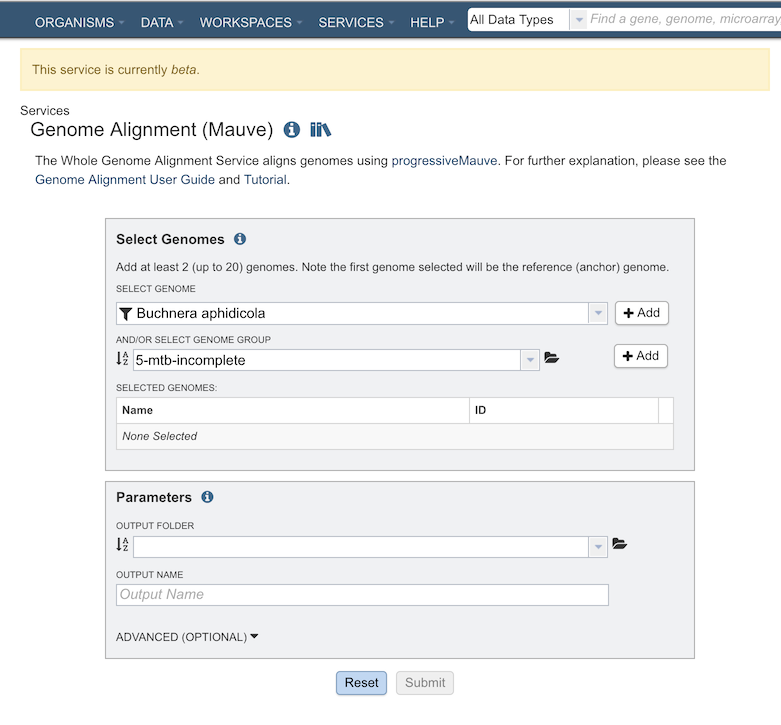
Select Genomes¶
Specifies the genomes (at least 2) to have aligned.
Select Genome¶
Genomes for inclusion in the ingroup for the tree. Type or select a genome name from the genome list. Use the “+ Add” button to add to the Selected Genome Table.
And/Or Select Genome Group¶
Option for including a genome group from the workspace. Can be included with, or instead of, the Selected Genomes.
Parameters¶
Output Folder¶
The workspace folder where results will be placed.
Output Name¶
Name used to uniquely identify results.
Advanced Options¶
Manually set seed weight: The seed size parameter sets the minimum weight of the seed pattern used to generate local multiple alignments (matches) during the first pass of anchoring the alignment. When aligning divergent genomes or aligning more genomes simultaneously, lower seed weights may provide better sensitivity. However, because Mauve also requires the matching seeds must to be unique in each genome, setting this value too low will reduce sensitivity.
Max gapped aligner length: Maximum number of base pairs to attempt aligning with the gapped aligner
Max breakpoint distance scale: Set the maximum weight scaling by breakpoint distance. Defaults to 0.9
Conservation distance: Scale conservation distances by this amount. Defaults to 1
Weight: Minimum pairwise LCB score
Min scaled penalty: Minimum breakpoint penalty after scaling the penalty by expected divergence
hmm-p-go-homologous: Probability of transitioning from the unrelated to the homologous state. Default is 0.0001
hmm-p-go-unrelated: Probability of transitioning from the homologous to the unrelated state. Default is 0.000001
Output Results¶
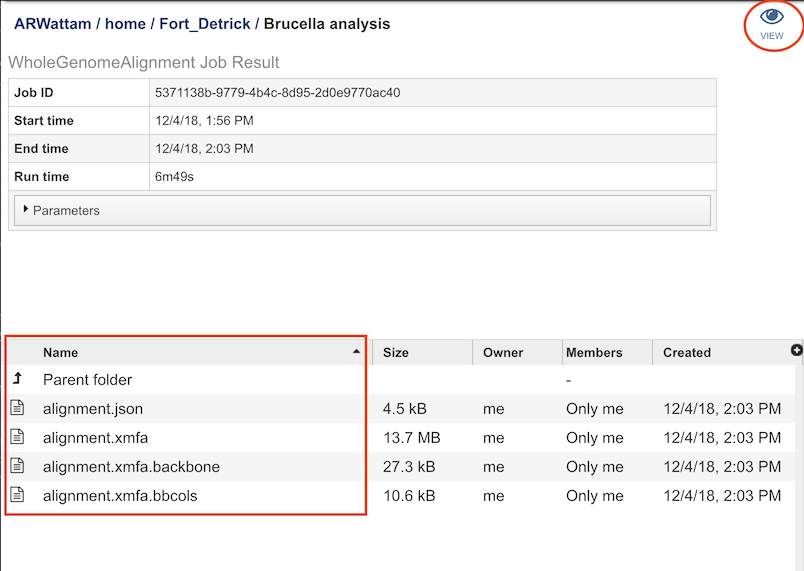
The Genome Alignment Service generates files that are deposited in the Private Workspace in the designated Output Folder. These include
alignment.xmfa - The Mauve alignment file.
alignment.json - The LCB coordinates in JSON format.
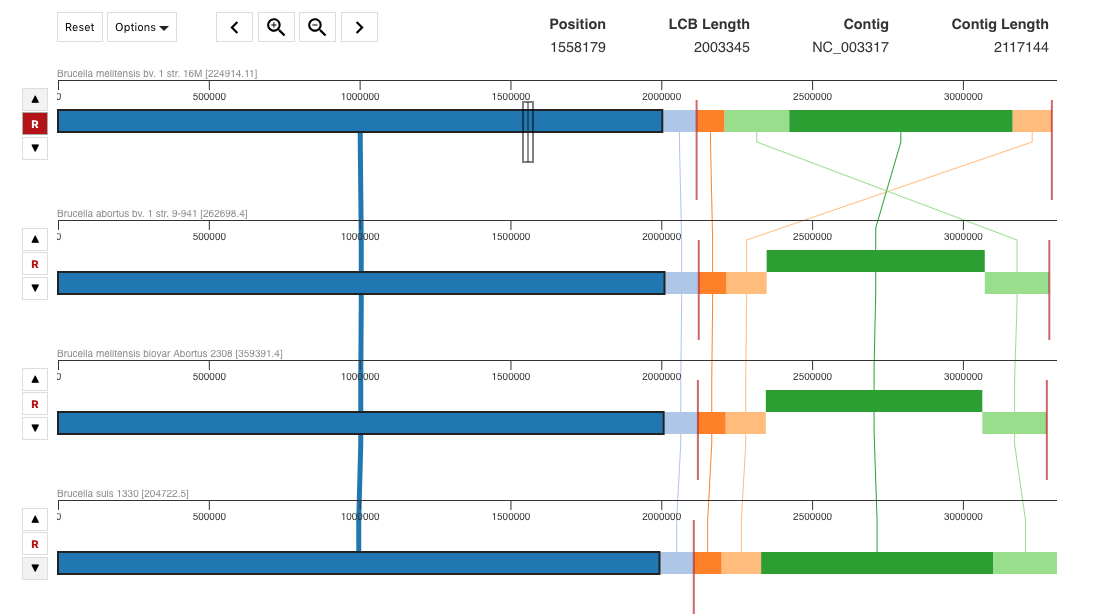
Clicking on “VIEW” at the top right of this page will bring will allow you to visualize the genome alignment:
References
Darling AE, Mau B, Perna NT (2010) progressiveMauve: Multiple Genome Alignment with Gene Gain, Loss and Rearrangement. PLOS ONE 5(6): e11147. https://doi.org/10.1371/journal.pone.0011147
http://darlinglab.org/mauve/user-guide/