Comprehensive Genome Analysis Service¶
The Comprehensive Genome Analysis Service is a streamlined analysis “meta-service” that accepts raw genome reads and performs a comprehensive analysis including assembly, annotation, identification of nearest neighbors, a basic comparative analysis that includes a subsystem summary, phylogenetic tree, and the features that distinguish the genome from its nearest neighbors.
Keywords: Genome analysis, Genome assembly, Genome annotation, Genome quality, Similar genomes, Comparative genomics, Phylogenetic tree, Genome Report.
Submitting a Comprehensive Genome Analysis job¶
Finding the service.
a. Click on the Services tab at the top of the page, and then click on Comprehensive Genome Analysis (CGA).
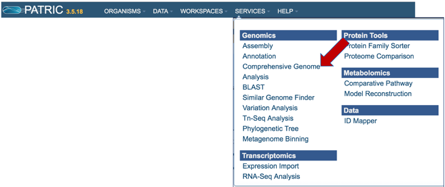
b. This will open the landing page for the service.
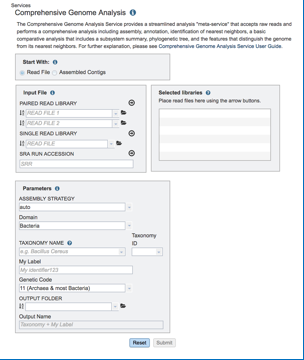
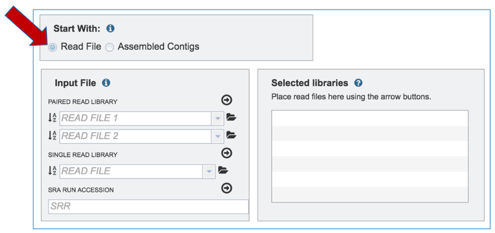
Input files: Uploading Reads or Contigs. The default setting is to analyze reads. PATRIC accepts reads ending in .fq, .fastq, .fa, .fasta, .fq.gz, .fastq.gz, .fa.gz, .fasta.gz
Input File-Uploading Paired End Reads.
a. To upload Paired End Reads, click on the Folder icon at the end of the text box underneath Paired Read Library. This will open a pop-up window. Click on the Upload icon in the upper right corner.
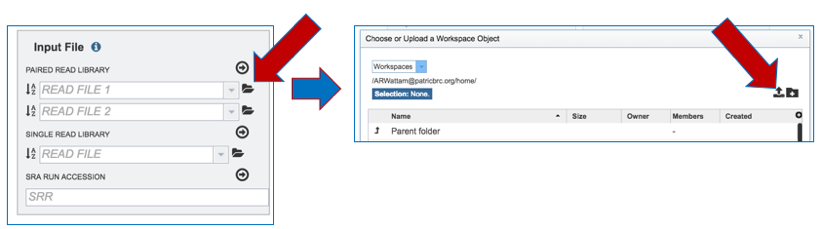
b. This will open a new pop-up window. Click on the blue bar that says Select File. This will open a pop-up window that gives access to files on your computer. Select the file of interest and click Open.
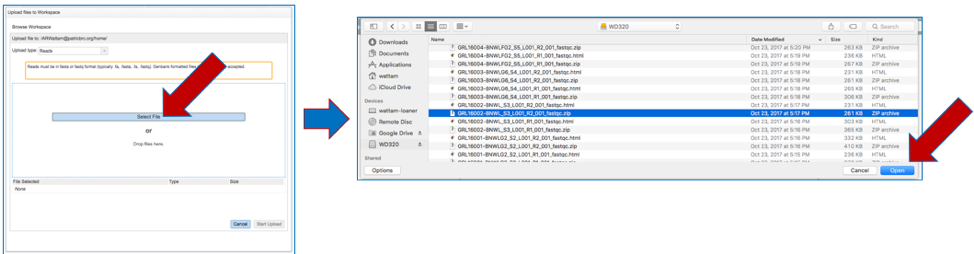
c. The name of the selected file will appear below the grey bar that says File Selected. Click on the Start upload button at the bottom right of the window. Follow the progress of the upload by examining the Upload monitor at the bottom right of the PATRIC page. Do not submit the job until the upload progress registers 100%.
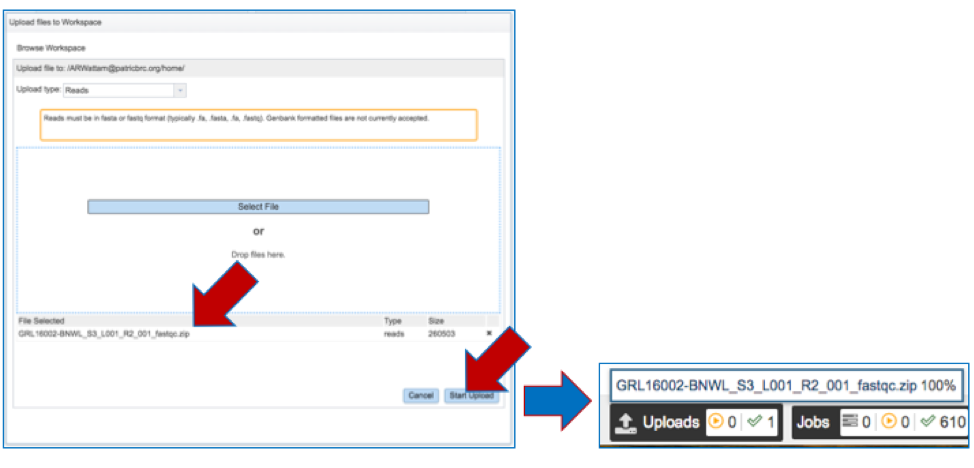
d. The name of the selected file will appear in the text box.
{width=”2.9444444444444446in” height=”2.031948818897638in”}
e. Repeat the process to upload the other read pair. Make sure that the NAMES OF THE FILES MATCH!
f. Next move the reads into the Selected libraries box. Click on the arrow above the Paired Read library text boxes. Doing this will move the paired end reads to the Selected libraries box.
{width=”6.5in” height=”1.4833333333333334in”}
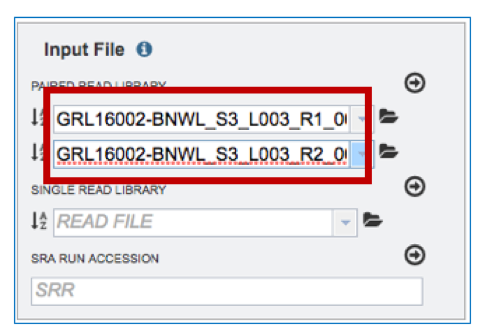
g. Ensure that the reads are correctly paired by clicking on the Information icon (i) in the Selected libraries text box.
{width=”6.5in” height=”1.9104166666666667in”}
h. If a mistake has been made, delete the incorrectly matched pairs by clicking on the Delete icon (x).
{width=”3.4in” height=”2.2547364391951006in”}
i. Some sequencing labs run several lanes from the same library during the sequencing process. The duplicate reads could be uploaded as well, if they exist, to create a more robust assembly. If available, repeat the process to upload more reads from the same strain.
Input File-Using previously uploaded Paired End Reads. To upload previously loaded Paired End Reads, click on the down arrow at the end of the text box under Paired Read Library. This will open a drop-down box that shows all the files that have been uploaded and tagged as reads. Clicking on one to select it will load it into the text box.
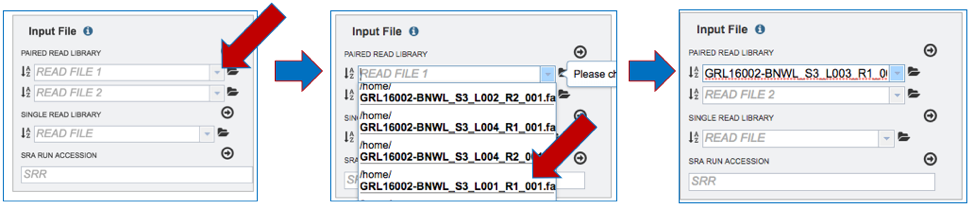 {width=”6.5in” height=”1.3972222222222221in”}
{width=”6.5in” height=”1.3972222222222221in”}
Input File-Single End Reads
c. To upload Single End Reads, follow the instructions listed above for Paired End reads, but upload the reads in the text box underneath Single Read Library.
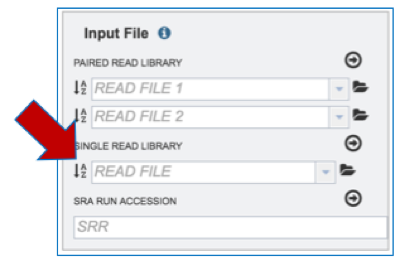
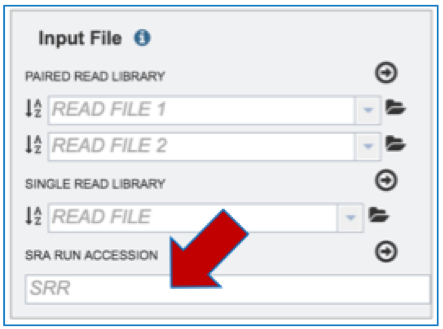
Input File-SRR numbers. To upload reads directly from the Sequence Read Archive[1], enter the SRR number into the text box underneath SRA Run Accession and follow the instructions listed above for Paired End reads.
 `
`Input File – Contigs. To upload Contigs, click on Assembled Contigs in the Start With box above Input File.
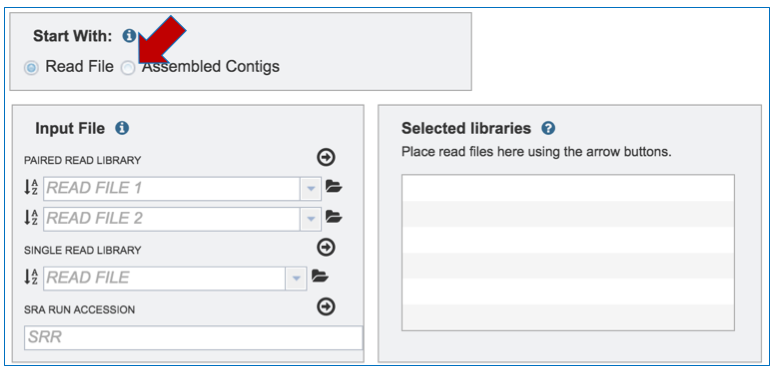
d. This will change the display, enabling uploading of contigs to the Input File. To upload Contigs, follow the instructions listed above for Paired End reads, but upload in the text box underneath Contigs.
`
Selecting Parameters for Comprehensive Genome Analysis
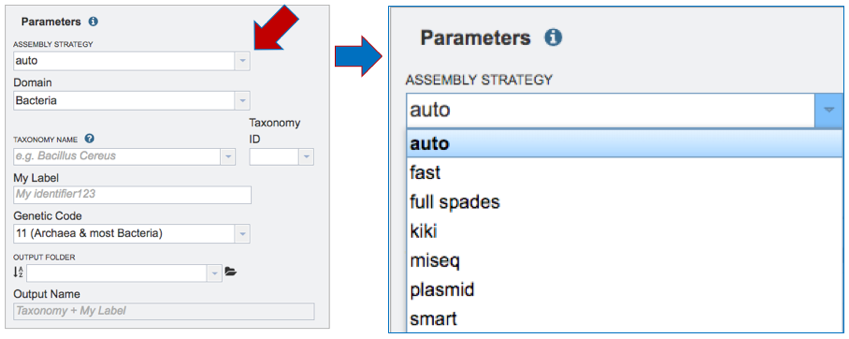
Assembly Strategy. PATRIC offers a number of assembly strategies that are listed below.
The auto assembly strategy runs BayesHammer[2] on short reads, followed by three assembly strategies that include Velvet[3], IDBA[4] and Spades[4], each of which is given an assembly score by ARAST, an in-house script.
The fast assembly strategy runs MEGAHIT[5] and Velvet, with each assembly given a score determined by ARAST.
Users can also choose the full spades strategy, which runs BayesHammer followed by Spades.
Choosing kiki runs the Kiki assembler, an in-house script.
Illumina MiSeq reads should be assembled using miseq, which runs Velvet with hash length 35, and then BayesHammer on reads and assembles with SPAdes with k up to 99, followed by a score using ARAST.
Plasmid runs BayesHammer on reads and assembles with plasmidSPAdes[6].
The smart strategy can be used for long or short reads. The strategy for short reads when using smart involves running BayesHammer on reads, KmerGenie[7] to choose hash-length for Velvet, followed by the same assembly strategy using Velvet, IDBA and Spades. Assemblies are sorted with an ALE score[8] and the two best assemblies are merged using GAM-NGS[9].
PacBio and Nanopore long reads only work with the auto and smart strategies. In either case, they are automatically assembled using Miniasm[10].
To select one of the strategies, click on the down arrow at the end of the text box under Assembly Strategy.
Domain. Select Bacteria or Archaea for the Domain that reflects the sequenced genome.
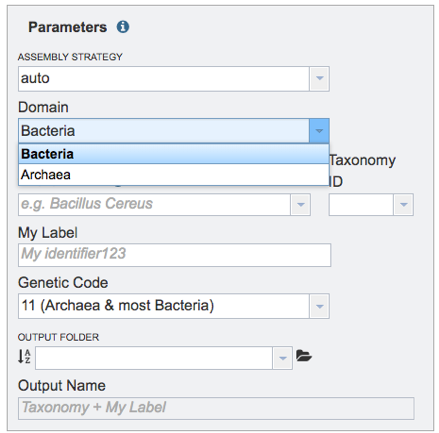
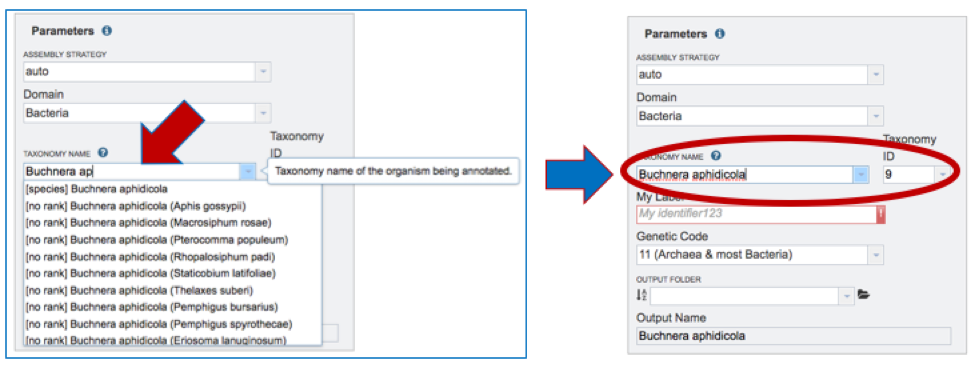
Taxonomy Name. Begin typing in the lowest ranked taxonomic name known for the sequenced isolate. It is best to be able to get to Genus, if possible. Once typing begins, a drop-down box will automatically appear with the taxonomic names in PATRIC that match the entered text. Click on the most appropriate name. This will fill the text box under Taxonomy Name with the selected name, and also include the Taxonomy ID. If the Taxonomy ID is known, that can be filled in and the ID and matching taxonomy name will be auto filled.
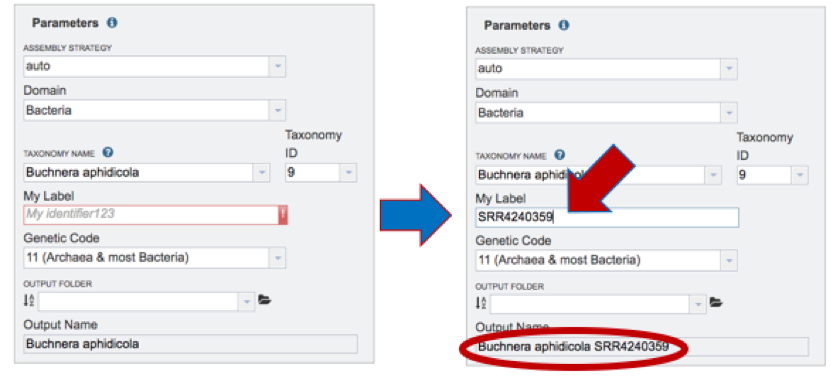
My Label. Give the genome a unique name by entering text in the box underneath My Label. The name that is entered will appear in the Output Name in the lowest text box.
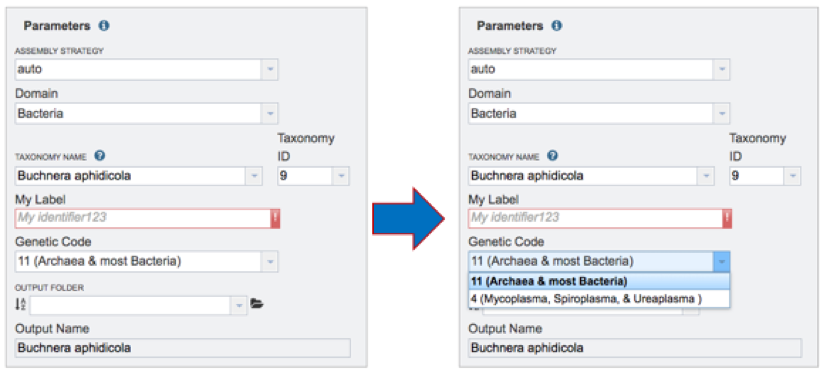
Genetic Code. Select the appropriate genetic code for the isolate.
Output Folder-Previously named. If a folder has been previously created, start typing the name in the text box underneath Output Folder, which will open a drop-down list of all folders in the workspace that match that text. Click on the appropriate folder.
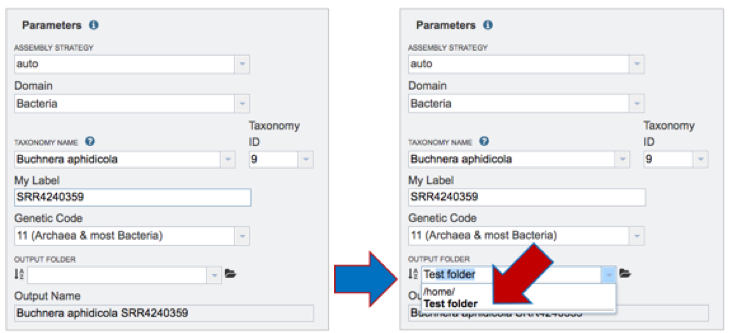
New Output Folder. To create a new folder for the job, click on the folder icon at the end of the text box underneath Output Folder. This will open a pop-up window. Click on the folder icon at the top right.
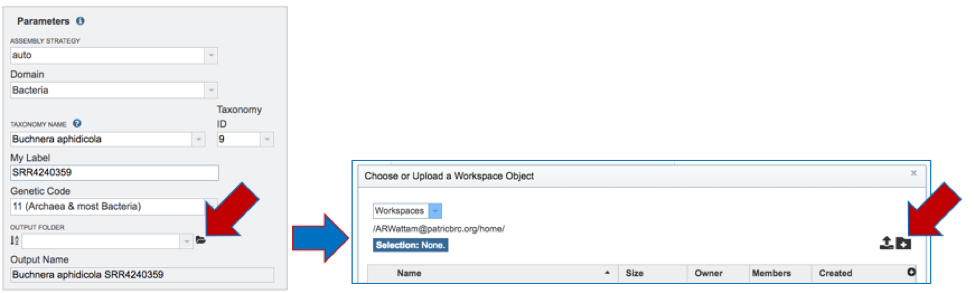
a. This will open a new pop-up window. Enter the name of the folder in the text box, and then click the Create Folder button.

b. The original pop-up window will appear. Find the name of the new folder, select it, and then click the OK button at the bottom right of the window. This will fill the name of the selected folder into the text box under Output Folder.

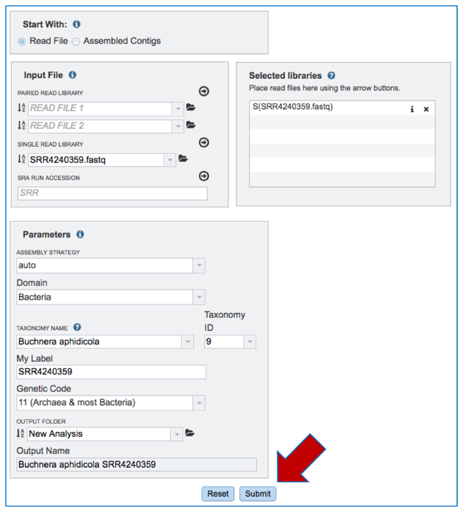
Submitting the Comprehensive Genome Analysis job
When all the parameters are entered correctly, the Submit button at the bottom of the page will turn blue. Click on that button, and the will enter the queue. You can monitor the progress on the Jobs page.
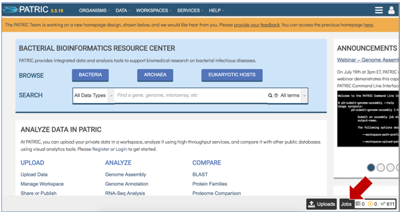
Finding the Comprehensive Genome Analysis job
Click on the Jobs monitor at the bottom right of any PATRIC page.
This will open the page where all jobs submitted to PATRIC are listed. Every Comprehensive Genome Analysis (CGA) job also launches an assembly and annotation job, which can be found imimagestely below the row that list the CGA job. To find out more information about the CGA job, click on the job of interest, and then on the view icon in the vertical green bar.
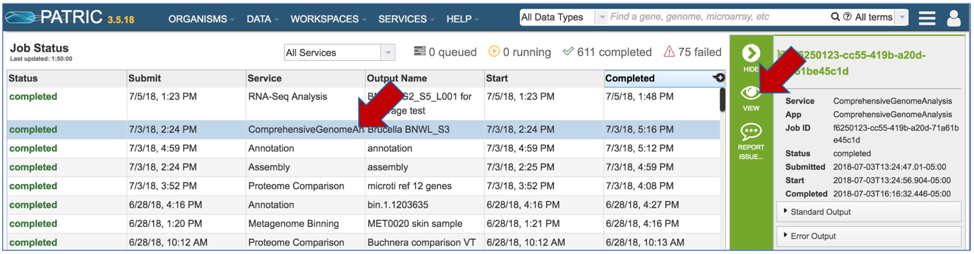
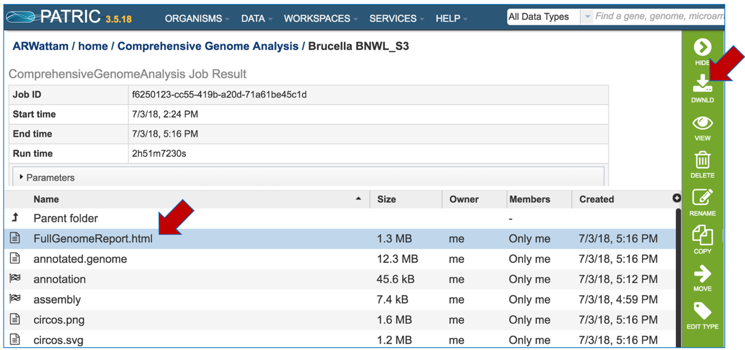
PATRIC now provides a genome announcement style document for any genome annotated using the Comprehensive Genome Analysis service. To see this document, select the row that contains the FullGenomeReport.html and click on the download icon in the vertical green bar.
Select an appropriate location on your computer and save the document, and then open it. You can view the document in any web browser
The full genome report provides a detailed summary of the genome. It begins with a summary of the genome quality, and then provides information for each step of the service, which includes assembly, annotation, and analysis of specialty genes and functional categories, and a phylogenetic tree of the new genome and its closest high-quality relatives.
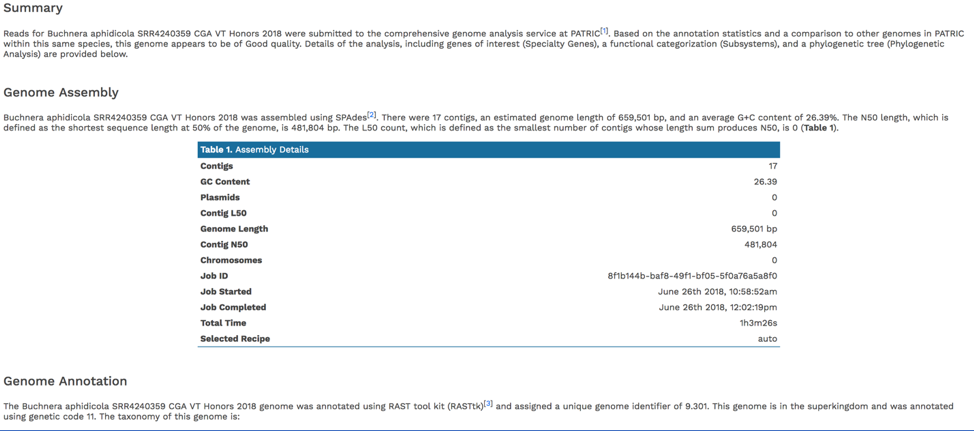
The summary will indicate is the genome is of good or poor quality.

Scrolling down to Genome Assembly will summarize the method selected for assembly and provide the statistics of interest. These statistics are those commonly provided when a genome is submitted as part of a publication.
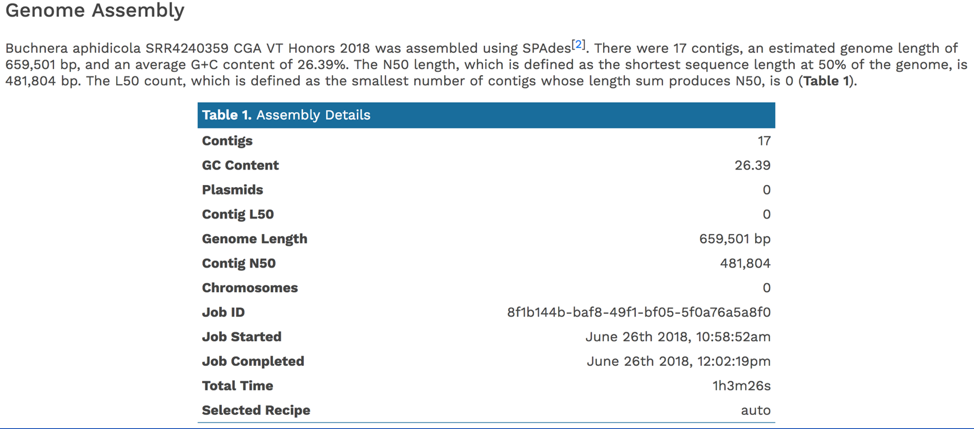
The Genome Annotation section describes the taxonomy of the genome, and genes and their functional divisions.
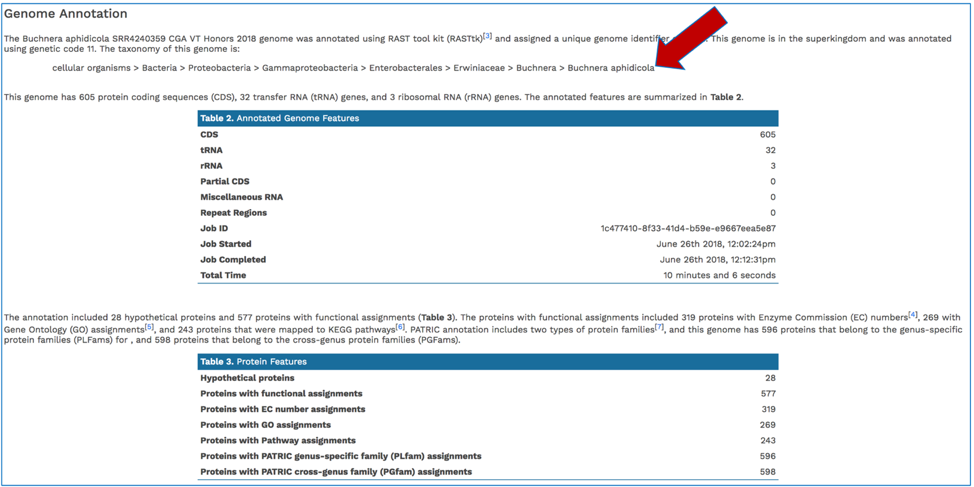
The Genome Annotation section also includes a circular diagram of the genes, their orientation, homology to AMR genes and virulence factors, and GC content and skew. Genes on the forward and reverse strands are colored based on the subsystem[11] that they belong to. A separate, downloadable svg or png of the circular graph image is available in the jobs list.
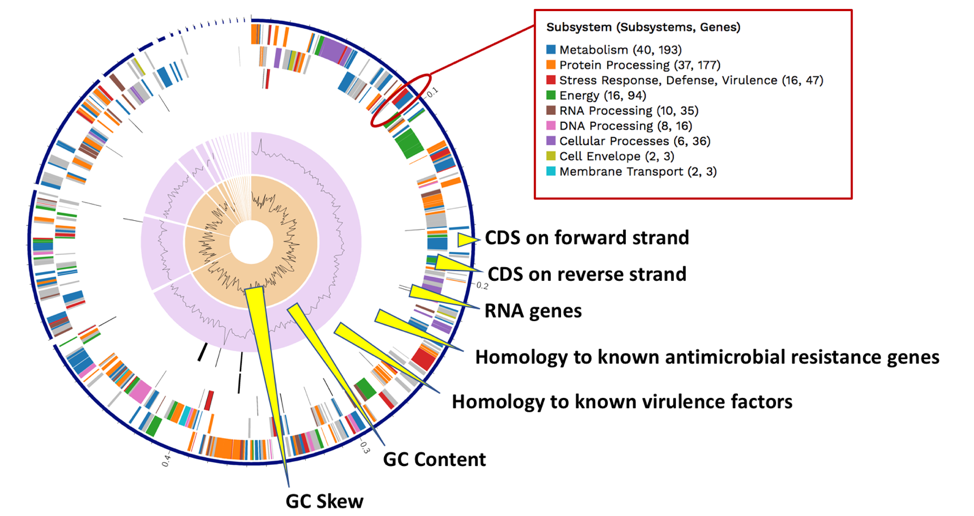
PATRIC BLASTs all genes in a new genome against specialty gene databases, including genes known to provide antibiotic resistance, virulence factors, and known drug targets. The CGA service shows the hits in the new genome have to those databases in a tabular form.
In addition, PATRIC provides a k-mer based detection method for antimicrobial resistance genes and shows the number of genes that share these k-mers.

PATRIC’s subsystem analysis identifies genes based on specific biological processes that they are hypothesized to be active in. The full genome report includes a pie chart showing the subsystems superclasses[11], and an indication of the number of subsystems within that superclass (first number) and the number of annotated genes that are part of the superclass (second number).

The CGA service identifies the closest relatives to the selected genome. It picks the closest reference and representative genomes using Mash/MinHash[12], and then takes twenty of PATRIC’s global protein families[13] that are shared across all the selected genomes to build a tree based on the amino acid and nucleotide alignments of those proteins, which are aligned using MUSCLE[14], and RaxML[15] is used to build the tree.. The genome submitted to the CGA is in red.
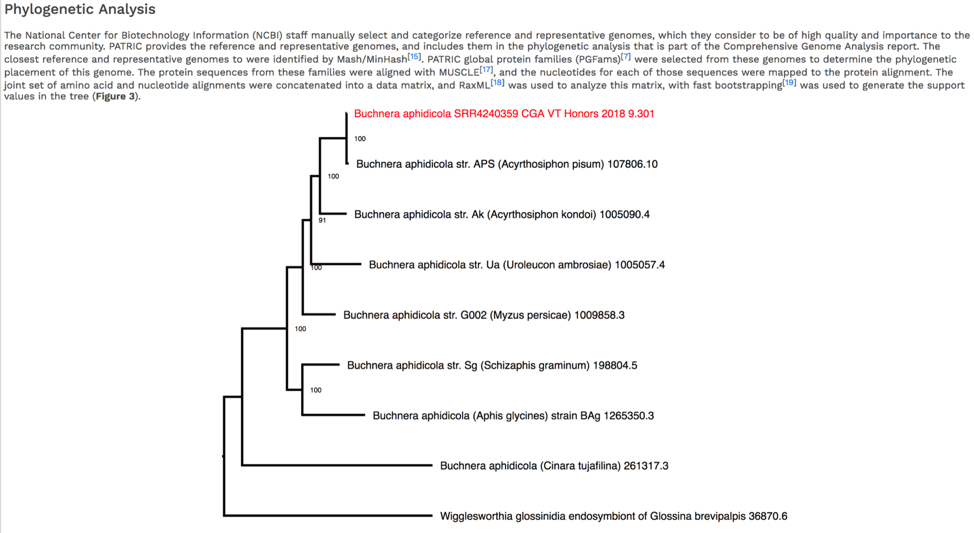
The newick file (.nwk) for the tree is available in the jobs list and can be used to construct the tree in another tree viewing program like FigTree (http://tree.bio.ed.ac.uk/software/figtree/) or the Interactive Tree of Life (https://itol.embl.de/).
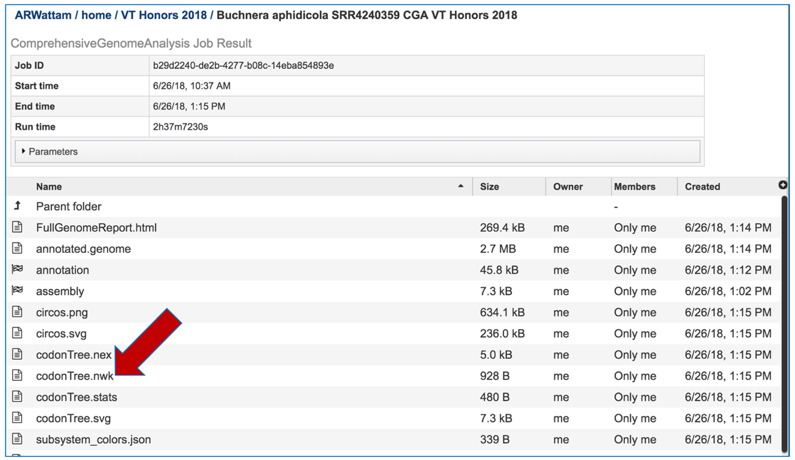
Viewing the genome in PATRIC
To view the integrated genome in PATRIC, double click on the row that has the flag icon and annotation in the landing page for the CGA job.
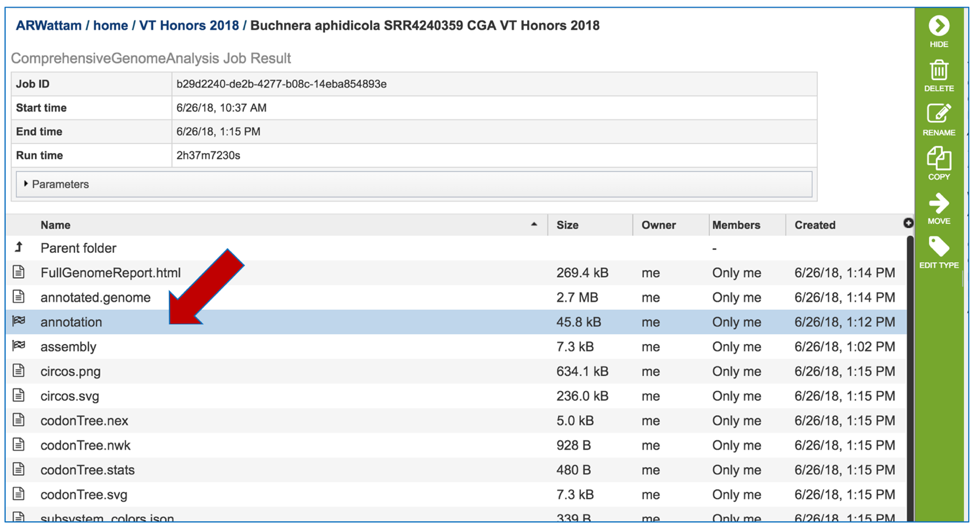
This will open a new page. Click on the view icon at the top right of the table.
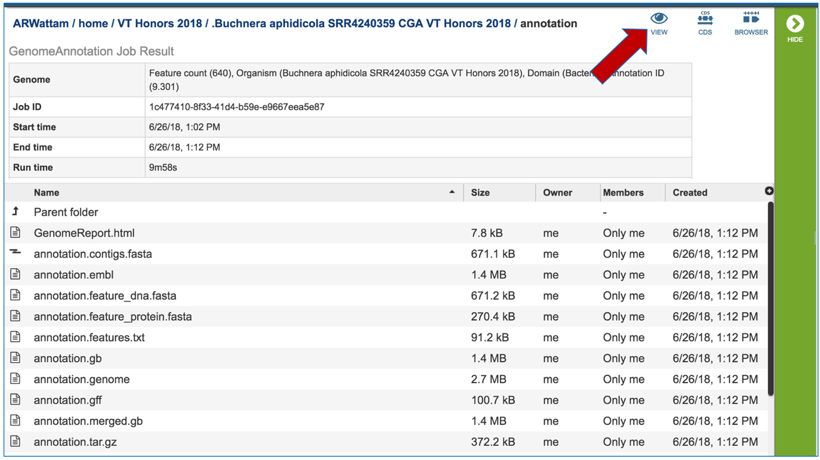
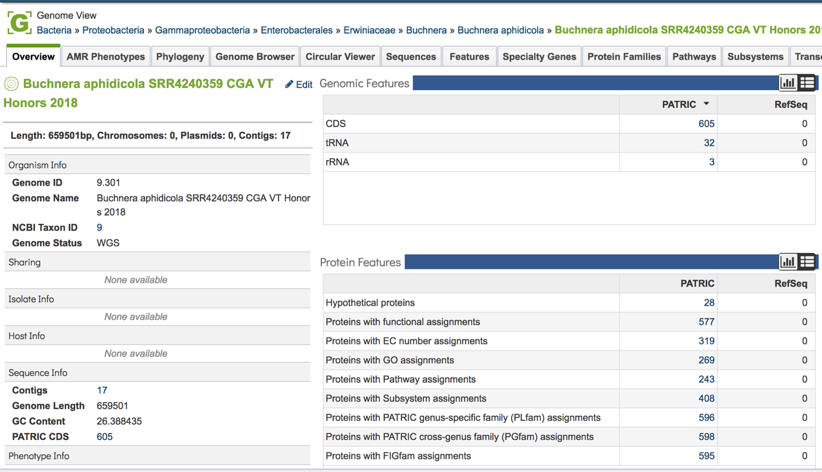
This will open the genome landing page for the genome that was assembled, annotated and analyzed using the CGA service.
References
Kodama, Y., M. Shumway, and R. Leinonen, The Sequence Read Archive: explosive growth of sequencing data. Nucleic acids research, 2012. 40(D1): p. D54-D56.
Nikolenko, S.I., A.I. Korobeynikov, and M.A. Alekseyev, BayesHammer: Bayesian clustering for error correction in single-cell sequencing. BMC genomics, 2013. 14(1): p. S7.
Zerbino, D.R. and E. Birney, Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome research, 2008. 18(5): p. 821-829.
Peng, Y., et al. IDBA–a practical iterative de Bruijn graph de novo assembler. in Annual international conference on research in computational molecular biology. 2010. Springer.
Li, D., et al., MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics, 2015. 31(10): p. 1674-1676.
Antipov, D., et al., plasmidSPAdes: assembling plasmids from whole genome sequencing data. bioRxiv, 2016: p. 048942.
Namiki, T., et al., MetaVelvet: an extension of Velvet assembler to de novo metagenome assembly from short sequence reads. Nucleic acids research, 2012. 40(20): p. e155-e155.
Clark, S., et al., ALE: a generic assembly likelihood evaluation framework for assessing the accuracy of genome and metagenome assemblies. Bioinformatics, 2013: p. bts723.
Vicedomini, R., et al., GAM-NGS: genomic assemblies merger for next generation sequencing. BMC bioinformatics, 2013. 14(7): p. S6.
Li, H., Minimap and miniasm: fast mapping and de novo assembly for noisy long sequences. Bioinformatics, 2016. 32(14): p. 2103-2110.
Overbeek, R., et al., The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res, 2014. 42(Database issue): p. D206-14.
Ondov, B.D., et al., Mash: fast genome and metagenome distance estimation using MinHash. Genome biology, 2016. 17(1): p. 132.
Davis, J.J., et al., PATtyFams: Protein Families for the Microbial Genomes in the PATRIC Database. Front Microbiol, 2016. 7: p. 118.
Edgar, R.C., MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic acids research, 2004. 32(5): p. 1792-1797.
Stamatakis, A., RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics, 2014. 30(9): p. 1312-1313.